

Elektroforeza kwasów nukleinowych
Z artykułu dowiesz się:
– Jak migrują kwasy nukleinowe w polu elektrycznym?
– Który bufor do elektroforezy DNA jest lepszy TAE czy TBE?
– Jaki żel do elektroforezy DNA jest lepszy agarozowy czy poliakrylamidowy?
– Jak wyznacza się wskaźnik EEO?
– Czy DNA / RNA widać w rozdziale elektroforetyczym?
– Jak określa się wielkość (długość) rozdzielanego DNA?
– Czy elektroforeza DNA jest techniką ilościową (PCR end point)?
– Na czym polega elektroforeza DNA w zmiennym (pulsowym) polu elektrycznym (Pulsed-field gel electrophoresis; PFGE)?
Elektroforeza kwasów nukleinowych jest łatwiejsza niż białek o której pisaliśmy tutaj -> Na czym polega elektroforeza białek? Czym jest izotachioforeza, rozdział SDS-Page, BN-Page i elektroforeza nieciągła (DISC)?
Jednak zjawiska towarzyszące obu aplikacjom są podobne. Przyjrzyjmy się im.
Jak migrują kwasy nukleinowe w polu elektrycznym?
Z definicji elektroforeza to zjawisko elektrokinetyczne, polegające na ruchu cząstek fazy rozproszonej w nieruchomej fazie rozpraszającej pod wpływem pola elektrycznego. Używając prostszego języka chodzi o rozdzielenie mieszaniny związków chemicznych (np. próbki DNA; lizatu z tkanki/komórek) na możliwie jednorodne frakcje, przez wymuszanie wędrówki ich cząsteczek w polu elektrycznym. Kwasy nukleinowe, dysocjują uzyskując jedynie ładunek ujemny (inaczej niż amfolityczne białka). W warunkach buforowych więc, po przyłożeniu pola elektrycznego poruszają się więc w kierunku elektrody dodatniej – anody.
Ich ruchliwość decyduje o szybkości migracji w żelu wg. wzoru
V= μ x E
gdzie,
V – to prędkość migracji [cm/s]
μ– to ruchliwość elektroforetyczna [cm2/V x s], zależna właśnie od ładunku, wielkości porów, pH buforu, lepkości, temperatury układu oraz siły jonowej.
E– to siła pola elektrycznego [V/cm], czyli siła z jaką pole elektryczne przesuwa cząsteczki – siłą napędową
Warto zauważyć, że im dłuższy żel [cm] tym napięcie [V] musi być większe, aby otrzymać taką samą wartość siły pola elektrycznego [V/cm] np. żel o długości 40 cm potrzebuje 10x więcej napięcia niż 10 cm mini żel.
Ponad to przepływ prądu jest proporcjonalny do przekroju poprzecznego żelu, czyli im grubszy i szerszy żel tym wyższy przepływ prądu. Z kolei wg wzoru moc to iloczyn napięcia i natężenia prądu (P= U x I [V x A]). Wynika z tego, że im dłuższy, szerszy i grubszy żel tym większą moc należy do niego przyłożyć oraz większe wydziela się tzw. ciepło dżulowe. Warto o tym pamiętać nastawiając parametry rozdziału, ponieważ temp. wpływa pozytywnie na ruchliwość elektroforetyczną cząsteczek.
Rozważając bardziej szczegółowo na wzrostruchliwości elektroforetycznej kwasów nukleinowych wpływają:
– bufor elektroforetyczny – ↑pH, ↑przewodnictwo elektryczne, ↑temperatura, ↓siła jonowa, ↓lepkość;
– ładunek cząsteczki (im silniej negatywnie naładowana cząsteczka tym szybciej migruje) – ↓ wartość pK (jeśli podobne do pH buforu), ↓ uwodnienie, ↓ otoczka jonowa (jeśli wokół kwasu nukleinowego stworzy się otoczka z przyciąganych kationów, obniża to ładunek ujemny cząsteczki);
– parametry rozdziału – ↓ wielkość porów w żelu (im mniejsze pory tym cząsteczkom trudniej się poruszać);
– rozmiar cząsteczki (im mniejsza cząsteczka tym szybciej migruje)- ↓ masa molekularna, ↓ wielkość, ↓ struktura (im bardziej nieregularna tym trudniej cząsteczce się poruszać);
– ↓rozmiar porów żelu (im większe pory żelu, tym cząsteczki szybciej migrują)
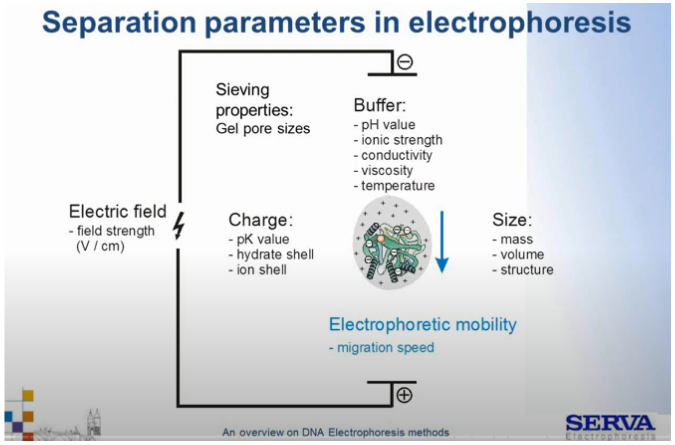
Fig1. Zestawienie parametrów warunkujących rozdział elektroforetyczny.
Najczęściej do elektroforezę kwasów nukleinowych wykorzystuje się bufor TAE lub TBE, zależnie od potrzeb.
Który bufor do elektroforezy DNA jest lepszy TAE czy TBE?
Oba bufory stosuje się powszechnie w laboratoriach. Warto jednak znać różnice między nimi, ponieważ wpływają one na odpowiednie ich dopasowanie zależnie od aplikacji. Ich nazwy kodują składowe-
– TBE (Tris-Borate-EDTA) rutynowo składa się z 89 mM Tris, 89 mM kwasu borowego i 2 mM EDTA (pH 8,3)
– TAE (Tris-Acetate-EDTA) z kolei zawiera 40 mM Tris, 20 mM kwasu octowego i 1 mM EDTA (pH 8,0)
Możemy zauważyć, że TBE ma wyższe pH co wpływa pozytywnie na ruchliwość cząsteczek i ich ładunek ujemny. Dlatego jest preferowany do elektroforezy DNA o wyższej rozdzielczości, szczególnie do oddzielania mniejszych fragmentów DNA (np. 100 bp do 1000 bp). Kwas borowy w TBE pomaga utrzymać stabilność pH podczas elektroforezy, co prowadzi do ostrzejszych prążków. Należy jednak pamiętać, że może on hamować aktywność enzymów poprzez tworzenie kompleksów z grupami hydroksylowymi w aminokwasach, także DNA jeśli nie będzie odpowiednio odpłukany od buforu (np. na kolumience krzemionkowej) nie będzie ulegać obróbce enzymatycznej. Bufor TAE jest powszechnie stosowany do rutynowej elektroforezy DNA i nadaje się do oddzielania większych fragmentów DNA (np. od 1 000 bp do 20 000 bp). Uważany jest także za mniej toksyczny i bezpieczniejszy w obsłudze niż buforu TBE, ponieważ nie zawiera kwasu borowego, który może być toksyczny w przypadku połknięcia lub wdychania. Zazwyczaj jest też tańszy.
Ponieważ, oba bufory różnie wpływają na ładunek cząsteczek, cząsteczki w nich rozdzielane będą inaczej migrować (porównaj Fig).
Zobacz nasz katalog produktów do elektroforezy DNA/RNA
Jaki żel do elektroforezy DNA jest lepszy agarozowy czy poliakrylamidowy?
Najczęściej do elektroforezy kwasów nukleinowych stosuje się agarozę, ale w pewnych wypadkach lepiej sięgnąć po poliakrylamid. Kiedy? Na przykład przy rozdzielaniu małych– kilku- kilkunastobazowych fragmentów z racji tego że żele te wykazują mniejszą elektroosmozę niż żele agarozowe oraz umożliwiają otrzymanie wystarczająco małych porów.
Regulując ilość czynnika sieciującego (C) do akrylamidu (T) wpływamy na wielkość przestrzeni w żelu. Im go więcej (%)tym bardziej usieciowany będzie żel i będą w nim mniejsze pory.
Np. 3% (C) i 5% (T) sprawia, że żel będzie miał pory 5 nm. Wartość nieosiągalna dla żelów agarozowych.
Sposób wykonywania obliczeń przedstawia Fig2.

Fig2. Zobrazowanie struktury porów żelu agarozowego i poliakrylamigowego.
Agaroza – polisacharyd, tworzy duże przestrzenie przez, które przenikają cząsteczki. Pory w 1% żelu agarozowym mają wielkość ok 150 nm. To dużo, pomimo tego jednak żele odznaczają się dużą odpornością mechaniczną, dzięki helikalnej strukturze ścian wokół poru. Możliwe jest przygotowanie żeli agarozowych o średnicy porów 500 nm w których będą rozdzielane kapsydy wirusowe.

Fig3. Dobór najlepszej agarozy do celów aplikacyjnych. Agarozy będą się różniły między sobą rozdzielczością, zwartością i EEO. Jeśli szukasz agarozy ekonomicznej i pasującej do różnych aplikacji polecamy Serva Agarose Wide Range. Jeśli liczy się dla Ciebie komfort pracy wybierz tabletki zawierające po 0,5 g agarozy (Agarose Serva Tablets). Dostępna jest też wersja z barwnikiem.
Zobacz nasz katalog produktów do elektroforezy DNA/RNA
Warto pamiętać, że w polu elektrycznym poruszają się wszystkie naładowane cząsteczki układu. Dotyczy to również żelu i aparatu do elektroforezy. Agaroza jest polisacharydem ekstrahowanym z agaru agaru pochodzącego z czerwonych wodorostów. Odczyszczana jest od agaropektyny, zawsze jednak trochę tego komponentu pozostaje wpływając na taki parametr odczynnika jak EEO (Elektro Endo Osmoza). Agaropektyna zawiera w sobie grupy siarczanowe i karboksylowe naładowane negatywnie w pH układu. Są one przyciągane przez anodę, racji jednak, na „uwięzienie” w sieci macierzy nie mogą migrować. Dochodzi do ruchu uprotonowanych cząsteczek wody w drugą stronę (tzw. kontreakcja), – tzw. przepływ elektroosmotycznym lub elektroendoosmoza. Zjawisko to może następować także wtedy, gdy rozdział przeprowadzany jest w szkle (kapilary), a nie w plastiku. Przyciągane są wówczas przez elektrodę grupy SiO– (Fig4.).
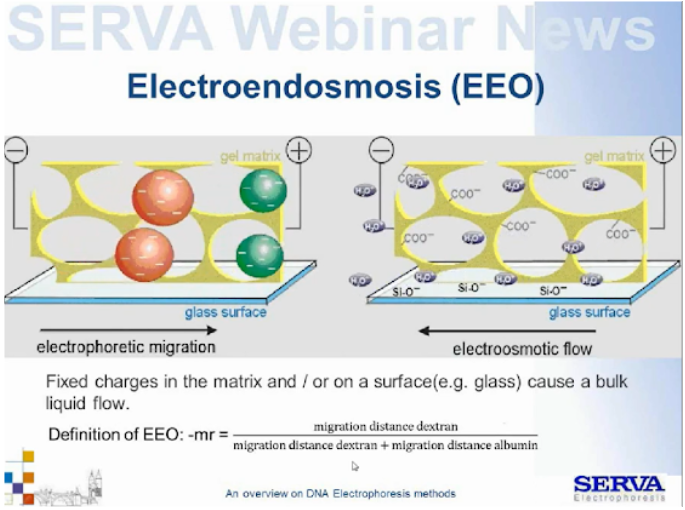
Fig4. Schematyczne przedstawienie migracji podczas przepływu elektroosmotycznego.
Wracając jednak do agarozy, te które mają najniższy wskaźnik EEO jakościowo są najlepszymi żelami agarozowymi do przeprowadzania elektroforezy. Również kosztują więcej. Wzmożona elektroendoosmoza może doprowadzić do powstawania zamazanych, mało ostrych prążków po wybarwieniu. Doprowadza także do zmniejszenia ilości wody w okolicach katody i „zalania” anody – dlatego elektroforezę kwasów nukleinowych przeprowadza się w poziomie, a żel jest całkowicie zatopiony/zanurzony w buforze, aby nie podsychał (submarine). Zazwyczaj przepływu elektroosmotycznego nie można zaobserwować, ponieważ ubytek medium jest kompensowany napływem nowego w zaprojektowanym układzie (cyrkulacja)


Fig5. Przykładowe aparaty do przeprowadzania elektroforezy poziomej.
Dowiedz się więcej o BlueMarine i MupidOne.
Zobacz nasz katalog produktów do elektroforezy DNA/RNA
Jak wyznacza się wskaźnik EEO?
Na żelu z testowanej agarozy przeprowadza się „rozdział” obojętnego ładunkowo dekstranu oraz albuminy. Ponieważ polimer nie ma ładunku, sam z siebie nie będzie poruszał się na żelu. Przesuwać go będzie przepływ elektro osmotyczny. Albumina jest wyznacznikiem prędkości migracji. Następnie dystans przebyty przez dekstran dzieli się przez sumę dystansów albuminy i dekstranu. Tak otrzymujemy EEO.
Czy DNA / RNA widać w rozdziale elektroforetyczym?
Cząsteczki kwasów nukleinowych na żelu nie są widoczne gołym okiem. Dlatego należy przeprowadzić ich wybarwianie. Można tego dokonać po rozdziale zanurzając żel w roztworze odpowiednich barwników, ale najczęściej dodaje się ich na etapie tworzenia żelu agarozowego. Do roztworu rozpuszczonej, ostudzonej agarozy w buforze (TAE,TBE) dodaje się odpowiednią objętość barwnika. Całość wylewa się na tackę, wkłada grzebień formujący dołki dla próbek i ewentualnie usuwa powstałe bąble powietrza. Barwniki czy to umieszczone wcześniej w żelu czy dostające się do niego podczas inkubacji w roztworze, interkalują pomiędzy nici kwasów nukleinowych. Po dostarczeniu im energii wzbudzenia (wzbudzeniu odpowiednią długością fali) oddają energię w postaci kwantu światła tj świecą. Sygnał ten możemy obserwować i zbierać za pomocą urządzeń rejestracyjnych (Fig6.).

Fig6. Przykładowe urządzenia pozwalające na rejestrację sygnału.
Zobacz nasz katalog produktów do elektroforezy DNA/RNA
W przeszłości najpopularniejszym barwnikiem stosowanym w tym celu był bromek etydyny (EtBr) . Ma on jednak silne właściwości kancerogenne, dlatego powoli wypierają bo bezpieczniejsze rozwiązania. W naszej ofercie możecie znaleźć tak samo czułe lub nawet czulsze Serva DNA Stain G i Serva DNA Clear Stain G o podobnych widmach ekscytacji i emisji co EtBr (odpowiednio Ex.300 Em. 530 nm. oraz Ex.295 nm Em.530 nm.) (Fig7,8.). Serva DNA Clear Stain G pozwalana uzyskanie bardziej wyraźnych prążków, ograniczenie tła i ma większą czułość. Drugi wymieniony barwnik jest opcją ekonomiczniejszą (Tab1). Czasami stosuje się także barwnik znany wcześniej jako SYBR Green.
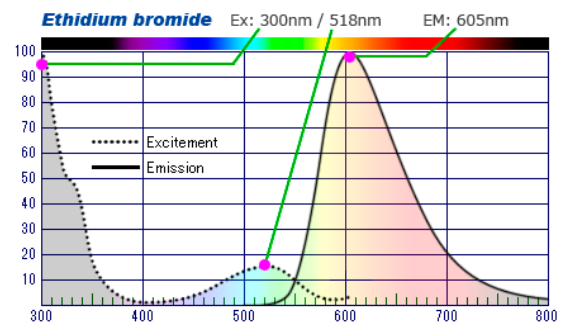
Fig7. Widma wzbudzenia i emisji dla bromku etydyny. Do wzbudzenie dochodzi najczęściej przy długościach fali odpowiadających UV, ale można go też wzbudzić falą o dł. 518 nm. Emisja osiąga swoje maximum intensywności przy długości 605 nm. Dla Serva DNA
[źródło mecanusa.com]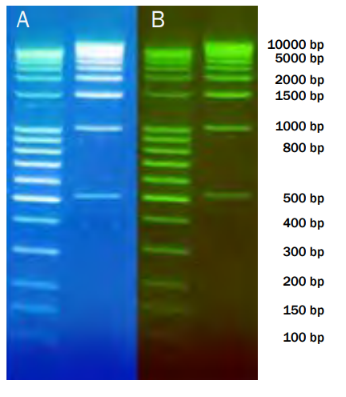
Fig8. Próbki DNA zostały rozdzielone w 1,5% żelu agarozowym. Do wstępnego barwienia rozcieńczono SERVA DNA Stain Clear G w stosunku 1:25 000. Barwienie wizualizowano za pomocą transiluminatora przy długości fali 312 nm. Linia 1: SERVA DNA Standard 100 Bp ladder, (nr kat. 39312). Linia 2: Drabinka SERVA DNA Standard 1KBp, (nr kat. 39314). Agaroza SERVA do elektroforezy DNA, (nr kat. 11404); Rozdział na aparacie BlueMarine™ 100, (nr kat. BM 100); 35 min, 150 V. A: bez filtra pomarańczowego; B: z filtrem pomarańczowym.
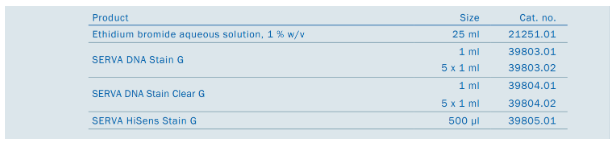
Tab.1
| Serva DNA Stain Clear G | Serva DNA Stain G | SERVA HiSens Stain G | |
| Certyfikaty bezpieczeństwa | Dostępne są następujące raporty potwierdzające bezpieczeństwo barwnika – Ames Test, Acute Oral Toxicity Test, Mouse Bone Marrow Micronucleus Test, Chromosome Aberration Test | Dostępne są następujące raporty potwierdzające bezpieczeństwo barwnika – Ames Test, test porównawczy z bromkiem etydyny (Ames), Mouse Bone Marrow Micronucleus Test, Chromosome Aberration Test | Dostępne są następujące raporty potwierdzające bezpieczeństwo barwnika – Ames Test, test porównawczy z bromkiem etydyny (Ames), test cytotoksyczności |
| Wydajność | 1 ml wystarcza na wybarwienie 17 – 25 L żelu agarozowego. | 1 ml wystarcza na wybarwienie 50 – 65 L żelu agarozowego. (proporcje dodawania 1:20.000 – 1:50.000) | 1 ml wystarcza na wybarwienie 10 L żelu agarozowego. |
| Cena | $$ /L | $/L | $$$/L |
| Maksimum wzbudzenia | 490 nm | 300 nm | 480 nm |
| Dodatkowe maksima wzbudzenia | 270, 290nm | ok. 450nm | 250 nm |
| Maksimum emisji | 530 nm | 530nm | 509 nm |
| Rozpuszczalność | rozpuszczalny w wodzie | rozpuszczalny w wodzie | rozpuszczalny w wodzie |
| Przechowywanie | przechowywanie w temperaturze pokojowej, chronić przed światłem | przechowywanie w temperaturze pokojowej, chronić przed światłem | przechowywać w zamrażarce, chronić przed światłem |
| Sposób użycia | do dozowania do płynnej agarozy lub wybarwianie po zakończeniu elektoroforezy (5-60 min inkubacja) | do dozowania do płynnej agarozy lub wybarwianie po zakończeniu elektoroforezy (5-60 min inkubacja) | do dozowania do płynnej agarozy lub wybarwianie po zakończeniu elektoroforezy (10-30 min inkubacja) |
| możliwość 2-3 krotnego wykorzystania roztworu barwnika do barwienia po zakończeniu elektroforezyNa 100 ml buforu 10-25 ul barwnika (żel grubości <0,5 cm) | możliwość 2-3 krotnego wykorzystania roztworu barwnika do barwienia po zakończeniu elektroforezy Na 100 ml buforu 5-15 ul barwnika (żel grubości <0,5 cm) | Na 100 ml buforu 10 ul barwnika (żel grubości <0,5 cm) | |
| w przypadku dodawania produktu do płynnej agarozy dodać 4-6 ul/100 ml roztw. agarozy (0,8-3%) | w przypadku dodawania produktu do płynnej agarozy dodać 1,5-2 ul/100 ml roztw. agarozy (0,8-3%) | w przypadku dodawania produktu do płynnej agarozy dodać 10 ul/100 ml roztw. agarozy (0,8-3%) | |
| Wyróżnik | Czysty obraz | Możliwa wizualizacja prążków w świetle widzialnym (>20 ng DNA) co pozwala na eliminację używania UV Ekonomia | Czułość (0,1 ng – fragmentu dsDNA dł. 4 kB) |
Zobacz nasz katalog produktów do elektroforezy DNA/RNA

Zapytaj nas o wygodnie porcjowaną agarozę w tabletkach z dodatkiem Serva Stain Clear G
Jak wspomniano wcześniej, nie można prowadzić obserwacji migracji DNA na żelu w czasie rzeczywistym (czyli bez umieszczenia pod UV). Można jednak prowadzić obserwację rozdziału elektroforetycznego w świetle widzialnym poprzez śledzenie migracji barwników dodawanych do buforu obciążającego do próbek. Bufor obciążający zazwyczaj zawiera substancję o dużej gęstości jako bazę, niedozwalającą na wypłynięcie próbki z dołka (glicerol/ficoll/sacharoza) i wspomniany kolorowy związek, ułatwiający nakładanie na żel. Barwniki te, posiadają ładunek pozwalając nam zgrubnie określić zaawansowanie migracji (Fig9.). Do najbardziej popularnych należą:
– Bromofenol Blue – niebieski, migruje na wysokości fragmentu 300 bp
– Ksylenocyjanol – ciemno niebieski, migruje na wysokości fragmentu 4 000 bp
– Oranż G – pomarańczowy, migruje na wysokości fragmentu 50 bp
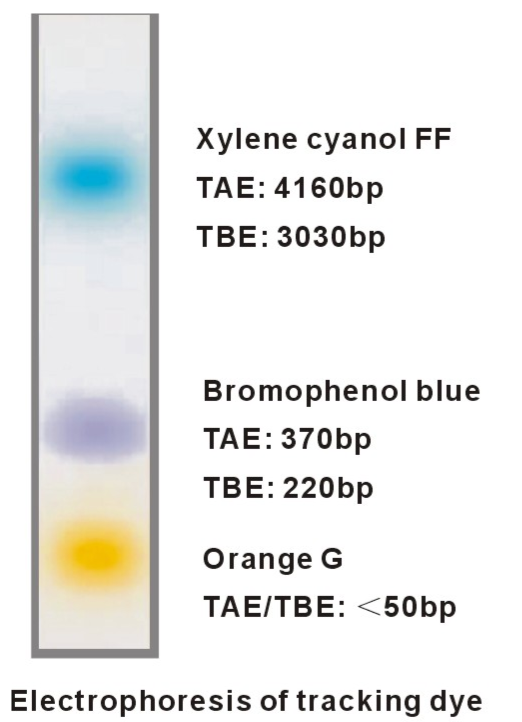
Fig9. Zobrazowanie migracji w żelu różnych barwników markerowych dodawanych do obciążników do próbek. Warto wybrać obciążnik, który nie będzie migrował na wysokości prążka, który chcemy następnie wyizolować z żelu, aby wyeliminować ewentualną inhibicję reakcji enzymatycznych.
[źródło gdsbio. com ]Często, premiksy do PCR (porównaj qPCR) zawierają już w sobie barwnik z obciążnikiem, aby ułatwić i przyspieszyć pracę podczas dokonywania późniejszej analizy na żelu. Należy do nich EmeraldAmp – szmaragdowy mix do PCR z polimerazą. Jego zielony kolor powstaje przez połączenie żółtego barwnika z niebieskim. Podczas migracji oba barwniki rozdzielają się. migrując w różnym tempie (wyznaczając ciężar ok 50 kb i ~3 kb). W ten sposób ułatwiają śledzenie postępu elektroforezy. Ponad to premiks zapewnia dużą swobodę w pracy z różnymi matrycami podczas rutynowego PCR i wysoką produktywność (#RR330A). Użyte w miksie barwniki nie przeszkadzają w przeprowadzaniu reakcji enzymatycznych, jak np. cięcie enzymami restrykcyjnymi.
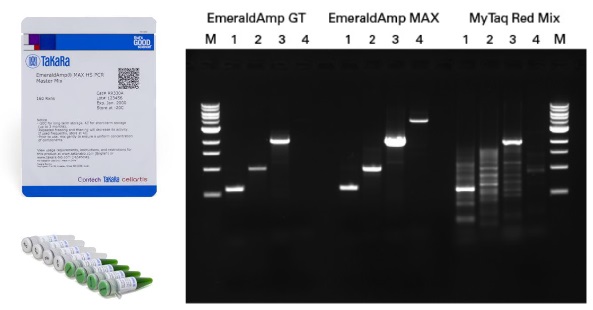
Zapytaj nas o próbkę EmeraldAmp!
Jak określa się wielkość (długość) rozdzielanego DNA?
Wyjaśniliśmy sobie jak śledzić postęp elektroforezy i że barwniki mogą być w tym przydatne. Pomimo iż możemy określać zakres w jakim migrują, nie możemy na ich podstawie precyzyjnie określić wielkości cząsteczki w nukleotydach; nt (base par; bp). Aby tego dokonać, podczas planowania elektroforezy zostawiamy dołek (czasem kilka) na aplikację markera molekularnego (zwanego też drabinką, ladder). Zawiera on mieszaninę fragmentów DNA lub RNA o znanych długościach. Ekstrapolując na podstawie migracji prążków drabinki i naszej próbki możemy określić bardziej precyzyjnie ciężar molekularny.
Warto także pamiętać, że DNA koliste (np. plazmidy) będą migrować inaczej niż cząsteczki liniowe. Możemy wyróżnić takie struktury jak CCC (covalently closed circular/ covalently bonded)- forma kolista, SP (supercoiled) – zwinięta forma kolista, OC (open circular/ nicked)- otwarto kolista i Lin (linear) – liniowa (Fig10.). Jeśli mamy taką możliwość, w celu rozstrzygnięcia, najlepiej rozdzielać takie cząsteczki ze standardem o wiadomej formie przestrzennej.
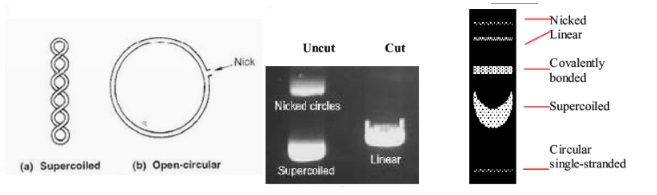
Fig10. Różnice w migracji plazmidów DNA zależnie od formy.
Czy elektroforeza DNA jest techniką ilościową (PCR end point)?
Odpowiadając krótko – nie. Czasem producenci określają stężenie fragmentów DNA drabinki (Fig11,12.), dzięki czemu zgrubnie możemy określić na podstawie intensywności sygnału ilość DNA w prążku. Często stosuje się także specjalne programy pomiaru densytometrycznego. Mimo wszystko nie otrzymamy w ten sposób, tak dokładnych danych o stężeniu jak w przypadku qPCR (porównaj qPCR), dlatego technika ta nie jest określana jako ilościowa.

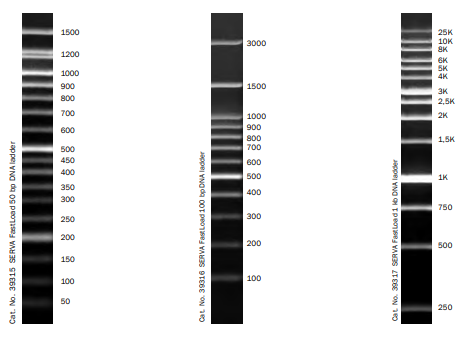
Fig11. Drabinki DNA (DNA Ladder/ DNA marker) dostępne w naszej ofercie.
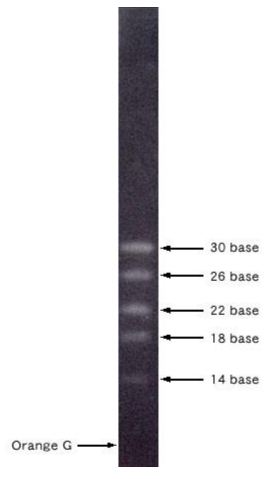
Fig12. Drabinka ssRNA (single strand RNA/RNA Ladder/ RNA marker) dostępna w naszej ofercie (#3416).
Zobacz nasz katalog produktów do elektroforezy DNA/RNA
Na czym polega elektroforeza DNA w zmiennym (pulsowym) polu elektrycznym (Pulsed-field gel electrophoresis; PFGE)?
Technika PFGE, czyli elektroforeza pulsacyjna, służy do rozdzielania cząsteczek DNA dużej wielkości. Pozwala na rozdzielenie cząsteczek o masie nawet powyżej 100 kB. Wykorzystuje się ją np. w mikrobiologii do tworzenia map restrykcyjnych szczepów bakterii. Dzięki analizie układu, wielkości i wysokości otrzymanych prążków można identyfikować szczepy bakteryjne i określać ich pochodzenie (Fig). Metoda ta jest szczególnie przydatna do typowania izolatów bakteryjnych z próbek klinicznych, takich jak krew, mocz i wymazy z ran, ale używa się jej także do śledzenia rozprzestrzeniania się antybiotykoodporności oraz wykrywania ognisk zakażeń bakteriologicznych żywności (Salmonella, Listeria). Elektroforeza pulsacyjna pozwoliła na identyfikację i śledzenie zakażeń szpitalnych wywołanych przez bakterie wielolekooporne np. ognisk opornego na metycylinę Staphylococcus aureus (MRSA) w szpitalach i placówkach opieki długoterminowej w Korei [Jung M.K. et all. Epidemiol Infect. 2014 Nov;142(11); doi: 10.1017 /S0950268813003543]. Z jej pomocą śledzi się przenoszenie kaset oporności na antybiotyki za pomocą plazmidów u szczepów Escherichia coli oraz Klebsiella pneumoniae.
Zobacz nasz wpis dotyczący antybiotyków jako narzędzi biotechnologicznych i broni w walce z chorobami.
Elektroforezę tę przeprowadza się na żelu agarozowym w buforze TBE dla lepszej rozdzielczości. Polega na naprzemiennym uruchamianiu elektrod w taki sposób, aby cząsteczka porusza się tzw. zygzakiem, skokowo. Technikę wymyślono ponieważ cząsteczki o masie powyżej 40 kB migrują z podobną, stałą szybkością. Wprowadzając pulsy niwelujemy to zjawisko. Większe cząsteczki mają mniejszą mobilność i są przez to mniej podatne na zmiany kierunku migracji (reorientację) niż mniejsze. Dzięki temu dochodzi do rozpętlenia cząsteczek a więc możliwy jest ich lepszy rozdział. Procedura zazwyczaj trwa długo od 16h do kilku dni.
We wspomnianym sporządzaniu map i typowaniu szczepów, przeprowadza się cięcie enzymami restrykcyjnymi przed PFGE. Każdy szczep różni się nieco pod względem układu nukleotydów, szczególnie w sekwencjach nie konserwowanych genetycznie. Zdarza się, że sekwencje, które są rozpoznawane przez enzym restrykcyjny w jednym szczepie, są już nie czytelne w innym w wyniku powstałej mutacji punktowej. W wyniku tego samego mechanizmu, mogą tworzyć się także nowe miejsca cięcia. Na tej podstawie można tworzyć dendrogramy pokrewieństwa szczepów, analizując układ prążków na żelu, rozdzielonych elektroforezą pulsacyjną. W internecie dostępne są symulacje cięcia genomów bakteryjnych enzymami restrykcyjnymi np. szeroko cytowane insilico.ehu. es/digest/.
Zapytaj nas enzymy restrykcyjne!
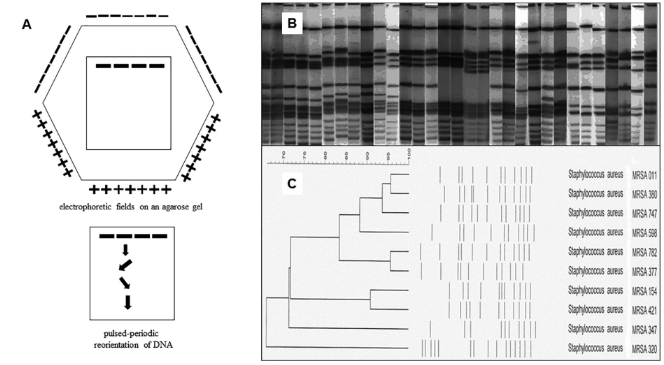
Fig13. Elektroforeza DNA w zmiennym (pulsowym) polu elektrycznym (Pulsed-field gel electrophoresis; PFGE), która pozwala na identyfikację szczepów Staphylococcus aureus A. Schemat rozmieszczenia elektrod w heksagonalnym układzie. Ważne, aby kąt pomiędzy pulsami był ok 110oC . Elektrody pracują pulsowo, naprzemiennie wymuszając zygzakowaty ruch cząsteczek. B. Wynik elektroforezy różnych szczepów Staphylococcus aureus C. Sporządzony na podstawie wyników elektroforezy dendrogram. Szczepy MRSA 011, MRSA 380 i MRSA 747 mają mniej niż 3 prążki różnicy w ich układzie, co określa bliskie pokrewieństwo szczepów. MRSA 347 i MRSA 320 mają więcej niż 7 prążków różnicy przez co uważane za szczepy niespokrewnione. [źródło Hui-min et. all. Infect Genet Evol. 2019 Oct:74:103935; DOI: 10.1016 /j.meegid.2019.103935]

