Na czym polega elektroforeza białek? Czym jest izotachioforeza, rozdział SDS-PAGE, BN-PAGE i elektroforeza nieciągła (DISC)?
22 Listopada 2023 (Ostatnia aktualizacja 24.10.2024)
Z artykułu dowiesz się:
– Czym jest elektroforeza i poznasz 4 zasadnicze rodzaje migracji zależne od warunków
– Czym jest elektroforeza strefowa, izotachioforeza, elektroforeza DISC, SDS-PAGE, elektroforeza natywna, blue native-PAGE
– Na czym polega sposób rozdziału pionowy i poziomy
– Czym jest elektroendoosmoza, ciepło dżulowe, efekt uśmiechu, efekt żaluzji weneckiej
– Czemu elektroforezę białek przeprowadza się na żelach poliakrylamidowych a nie agarozowych
Historycznie jako pierwszy elektroforezy użył w 1908 Karl Landsteiner do oddzielenia białek z surowicy krwi. Aktualnie jest stosowana w badaniach nad białkami, kwasami nukleinowymi, aminokwasami, cukrami, witaminami, hormonami, enzymami, lekami i innymi związkami chemicznymi.
Elektroforeza to technika analityczna i rzadziej preparatywna, stosowana w chemii i biologii molekularnej, np. genetyce.
Czym jest elektroforeza?
Zanim przejdziemy do elektroforezy białek. Wyjaśnijmy najpierw czym w ogóle jest elektroforeza.
Wyjaśnienie i zrozumienie zjawisk towarzyszących, może pomóc rozwiązać napotkane problemy.
Według definicji elektroforeza to zjawisko elektrokinetyczne, polegające na ruchu cząstek fazy rozproszonej w nieruchomej fazie rozpraszającej pod wpływem pola elektrycznego. Używając prostszego języka chodzi o rozdzielenie mieszaniny związków chemicznych (np. próbki DNA; lizatu z tkanki/komórek) na możliwie jednorodne frakcje przez wymuszanie wędrówki ich cząsteczek w polu elektrycznym. Cząsteczki różnych substancji różnią się zwykle ruchliwością elektroforetyczną. Parametr ten jest w przybliżeniu wprost proporcjonalny do ładunku elektrycznego cząsteczki i odwrotnie proporcjonalny do jej wielkości. Zależy także od kształtu cząsteczki.
Prędkość migracji (V; [cm/s]) można wyrazić wzorem:
V= μ x E
gdzie,
μ– to ruchliwość elektroforetyczna [cm2/V x s], zależna właśnie od ładunku, wielkości porów, pH buforu, lepkości, temperatury układu oraz siły jonowej.
E– to siła pola elektrycznego [V/cm], czyli siła z jaką pole elektryczne przesuwa cząsteczki.
Warto zauważyć, że im dłuższy żel [cm] tym napięcie [V] musi być większe, aby otrzymać taką samą wartość siły pola elektrycznego [V/cm]. Ponad to przepływ prądu jest proporcjonalny do przekroju poprzecznego żelu, czyli im grubszy i szerszy żel tym wyższy przepływ prądu. Z kolei wg wzoru moc to iloczyn napięcia i natężenia prądu (P= U x I [V x A]). Wynika z tego, że im dłuższy, szerszy i grubszy żel tym większą moc należy do niego przyłożyć oraz większe wydziela się tzw. ciepło dżulowe. Warto o tym pamiętać nastawiając parametry rozdziału.
Ważność tych parametrów np. ciepła dżulowego, będzie omówiona w dalszej części artykułu.
Nie należy także zapominać, że migracji cząsteczek ujemnie naładowanych w kierunku anody towarzyszy także przeciwstawny ruch cząsteczek dodatnie naładowanych. Uprotonowane cząsteczki wody oraz inne dodatnio naładowane grupy będą przyciągane w stronę elektrody ujemnej – katody. Ruch taki nazywa się przepływem elektroosmotycznym lub elektroendoosmozą. Zjawisko to może powodować rozmycie prążków, ich niewyraźne granice oraz wystąpienie innych anomalnych obrazów. Jest to zjawisko szczególnie widoczne gdy, używa się odczynników słabej jakości np. starego lub złej jakości akrylamidu. Kwas akrylowy – który uczestniczy w polimeryzacji, grupy karboksylowe, sulfonowe w zasadowym pH zostaną odprotonowane, zyskując ładunek ujemny, będą chciały migrować w kierunku anody, jednak ze względu na to, że są związane w żelu nie mogą. Dochodzi do przeciw reakcji, w wyniku której migracji ulegają uprotonowane cząsteczki wody. Dzieje się tak także gdy powierzchnie nie są elektrycznie obojętne np. może dochodzić do przyciągania tlenku krzemu ze szkła (Fig1).
Problem ten występuje także podczas używania żelów agarozowych. Jest nawet powszechniejsza, o czym później.
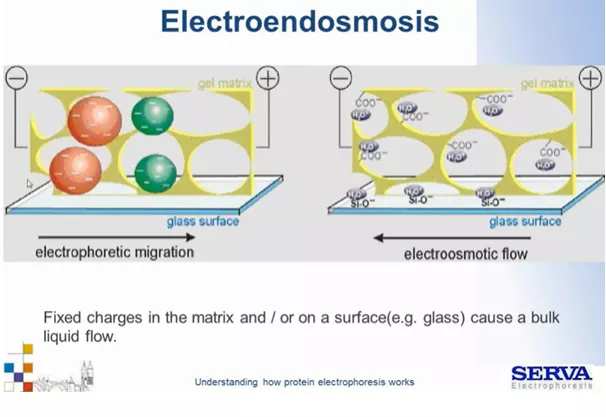
Fig1.
Schemat ukazujący zjawisko elektroendoosmozy. Szczegóły w tekście.[źródło: Serva]
Przechodząc do elektroforezy białek, można wyróżnić 4 zasadnicze rodzaje migracji, zależnie od zapewnionych warunków (Fig2):
– Elektroforeza strefowa, (potocznie rozumianą jako elektroforeza) – za jej pomocą można oddzielić mieszaninę naładowanych np. białek, peptydów, np. w żelu zapewniając migrację w stałym polu elektrycznym w oparciu o wielkość i ładunek cząsteczek.
– Elektroforeza fazy ruchomej – pierwszy zastosowany rozdział elektroforetyczny. Odbył się przy użyciu rurki w kształcie litery U wypełnionej buforem. Na dno nałożono mieszaninę białek Dodatnio naładowane cząsteczki zaczęły migrować w kierunku katody, a ujemnie naładowane w kierunku anody. Jak wydać na Fig.2 otrzymujemy mieszane strefy i niską rozdzielczością. To trudna technika więc ostatecznie przełączono się na elektroforezę strefową. Nie będzie tu szerzej omawiana.
– Izotachoforeza – (isotachiophoresis; ITP) z greckiego „z równą szybkością migracji” to proces rozdzielania jonów, które w polu elektrycznym przy zastosowaniu odpowiedniego układu buforowego (elektrolitu wiodącego i końcowego), formują się w strefy według ich zmniejszającej się ruchliwości i poruszają się z jednakową prędkością. W tej technice osiąga się migrację wszystkich różnych jonów z dokładnie taką samą prędkością.
– Ogniskowanie izoelektryczne (IEF) – inaczej izoelektrofokusowanie, to technika, która może być wykonywana wyłącznie ze związkami amfoterycznymi, czyli posiadającymi w cząsteczce grupy o charakterze kwasowym i zasadowym. Nie będzie szerzej omawiana w tym artykule.
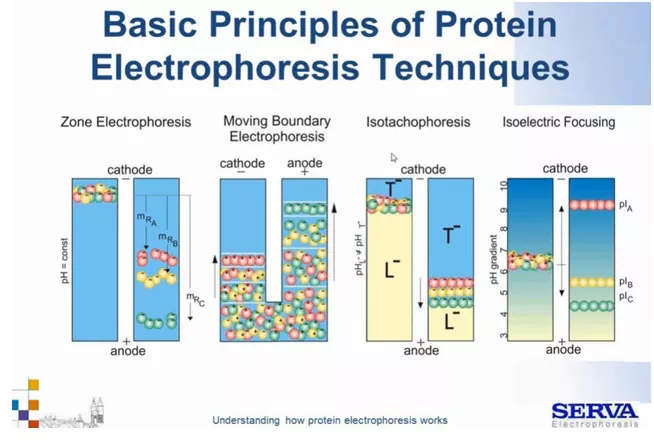
Fig2.
Schemat podstawowych technik elektroforezy białkowej. Kolory kulek oznaczają poszczególne frakcje białek, zaś plusy i minusy ich naładowanie. Kulki zielone oznaczają małe rozmiarowo cząsteczki, czerwone duże a żółte średnie. Więcej szczegółów w odpowiednich rozdziałach. [źródło: Serva]
Elektroforeza strefowa i izotachioforeza białek
Czym jest elektroforeza strefowa?
Jak wcześniej wspomniano jest potocznie rozumiana jako elektroforeza. Pozwala na rozdzielenie mieszaniny naładowanych cząsteczek np. białek, peptydów. Odbywa się w buforze o zasadowym, wysokim pH, który wymusza na cząsteczkach oddanie protonów (H+), w wyniku czego otrzymują netto ładunek ujemny. W przyłożonym polu elektrycznym, cząsteczki będą migrować do elektrody dodatniej -anody. Ten rodzaj elektroforezy można wykonywać tylko z wykorzystaniem faz ciekłych np. w kapilarach lub podczas swobodnego przepływu, kiedy stosuje się dodatkowo warstwę buforową lub żele agarozowe lub poliakrylamidowe. Te ostatnie stanowią pewną barierę fizyczną, generując opóźnienia w migracji zależnie od rozmiaru cząsteczek. W takich warunkach rozdział będzie się odbywał w oparciu nie tylko o ładunek, ale też o wielkość cząstek. Najwolniej będą migrować duże cząsteczki najmniej ujemne najszybciej zaś małe o silnym ujemnym ładunku. Wzdłużnie powstaną więc odrębne strefy, stąd nazwa – elektroforeza strefowa. Można ją przeprowadzać za równo w układzie pionowym jak i poziomym.
Na czym polega elektroforeza pionowa?
Elektroforezę białek przeprowadza się głównie w układzie pionowym, na żelach akrylamidowych, w systemach z żelem umieszczonym pomiędzy dwoma szybkami. Takie żele można własnoręcznie wylać, formując miejsca na próbki za pomocą plastikowego „grzebienia”. Żel zastygając formuje macierz z dołkami. W układzie znajduje się ponad to zbiornik górny i dolny. Próbka przepływa z góry na dół w układzie pionowym (wertykalnym).
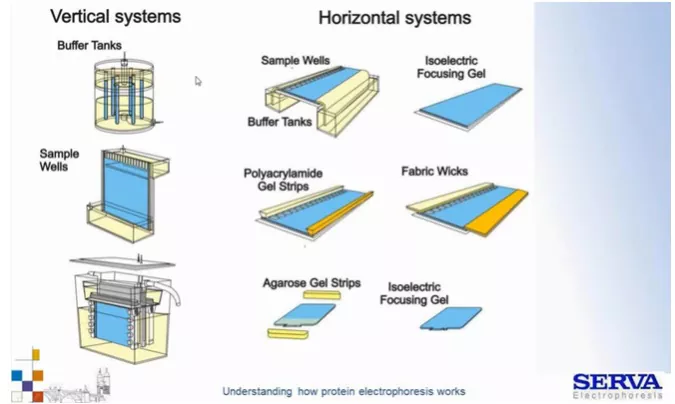
Fig 3.
Przykłady różnych układów (pionowe/poziome) rozdziałów elektroforetycznych. Przyjęło się, że w układzie pionowym (po lewo) rozdziela się białka natomiast poziomym kwasy nukleinowe (po prawo). Historycznie na początku rozdziały przeprowadzano w rurkach wypełnionych poliakrylamidem. Ruch następował wzdłuż rurek w dół. Na górze i dole znajdowały się zbiorniki, w których umieszczone były elektrody. Dalsze manipulacje rurkami były jednak nieporęcznie. Z czasem zaczęto stosować szybki pomiędzy, którymi znajdował się żel. Przykładem takiego systemu jest BlueVertical Prime Serva. Po prawej z kolei układy rozdziału poziomego (klasycznego rozdziału i IEF) z różnym zorganizowaniem układu buforowego. Na górze klasyczne duże zbiorniki z buforem, niżej bibuły/paski nasączone buforami oraz paski żelowe zawierające odpowiednie bufory. Przykładem takiego systemu jest HPE BlueHorizon Serva. Systemy te pozwalają na zmniejszenie ilości pracy związanej z buforami i pozwala na rozdział wielu próbek na raz. System posiada także chłodzącą szufladę chroniącą przed efektami uwalniania ciepła dżulowego. [źródło: Serva]


Zobacz katalog BlueVertical Prime I HPE BlueHorizon
Zjawisku temu towarzyszy wydzielanie się energii w postaci ciepła tzw. ciepła dżulowego. Wypromieniowywanie ciepła jest takie same w każdym punkcie wylanego żelu, jednak ze względu na to że krawędzie mają kontakt z większą ilością buforu, w związku z czym mogą efektywniej oddać ciepło, są chłodniejsze. Podwyższenie temperatury następuje w centrum żelu, co sprzyja powstawaniu łuku/ uśmiechu. Dzieje się tak ponieważ ciepło sprzyja przyspieszeniu migracji cząstek. Aby ograniczyć to zjawisko i móc np. porównywać próbki między sobą, stosuje się różnego rodzaju wymienniki cieplne – pompki wodne, wkłady chłodzące oraz zwiększa się objętość pojemnika buforowego mającego za zadanie również buforować/odprowadzać ciepło. Innym zjawiskiem o którym warto pamiętać to tzw. efekt żaluzji weneckiej. W związku z układem zbiorników i nagromadzaniu się ciepła, szybciej migrują białka z przodu żelu (w więc mniejsze/silniej naładowane) niż z tylu. Jest to szczególnie dobrze widoczne, gdy przyłożymy dużą moc.
Oba te efekty (efekt uśmiechu i efekt żaluzji weneckiej) możemy zaobserwować w rozdziale pionowym. Zwykle stosowanie dużego pojemnika anodowego, który chłodzi szybki a w konsekwencji żel, rozwiązuje te trudności. Wszystkie systemy pionowe pozwalają na rozdzielanie próbek w systemach zamkniętych.
Inaczej niż w przypadku rozdziałów poziomych.
Na czym polega elektroforeza pozioma?
Podczas elektroforezy poziomej żele nie są zamknięte pomiędzy płytkami i mogą ale nie muszą być zanurzone w buforze. Mogą też być lekko przykryte przez bufor a odbieranie ciepła zachodzi poprzez dodatkowy system chłodzący. W tym przypadku chłodzony jest bezpośrednio żel a nie bufor. Jak wspomniano wcześniej taki układ stosuje się głównie podczas elektroforezy kwasów nukleinowych oraz podczas ogniskowania izoelektrycznego białek. Ten rodzaj rozdziału daje dużo możliwości – można stosować klasyczne zbiorniki buforowe jak wspomniano wcześniej ale można np. stosować jedynie bibuły nasączone odpowiednimi buforami lub zamiast całych żeli stosować jedynie paski żelowe nasączone mocno zatężonym buforem.
Czym jest izotachioforeza?
Opisane powyżej sposoby rozdziału najczęściej występują w układzie elektroforezy strefowej – z rozdziałem migrujących frakcji białek zależnie od ich wielkości i naładowania, ale nie jest to regułą.
Podczas izotachioforezy cząsteczki poruszają się z tą samą prędkością jak wspomniano wcześniej. Aby do tego doszło potrzebny jest nieciągły układ, złożony najczęściej z dwóch buforów, w którym jony występują w zadanych kolejnościach. Technika pozwala na rozdział osobno, albo tylko kationów albo tylko anionów. Rodzaj rozdzielanych cząsteczek determinuje dobór składników buforów. Przy izotachioforezie białek będzie to rozdział anionów.
W żelu będzie występował jon/ elektrolit wiodący L– (tzw. leading) o większej ruchliwości od cząsteczek próbki np. Cl– z układu Tris-HCl (pH 6,8) a w buforze (running buffer) jon/elektrolit terminujący T– np. glicyna z układu Tris-Glycine o niskiej ruchliwości. W przypadku glicyny (amfolitu) pH układu 6,8 sprawia, że jest ona blisko swojego punktu izoelektrycznego a więc jest praktycznie obojętna elektrycznie. Próbka składa się ze związków o ruchliwości mniejszej od Cl i większej niż glicyna, w wyniku nałożenia próbki pomiędzy elektrolity, wszystkie cząsteczki migrują z tą samą prędkością mino różnej ruchliwości. Dzieje się tak w wyniku niwelowania powstawania tzw. „luki jonowej”. Małe, silnie naładowane, bardzo ruchliwe jony Cl– mają potencjał do szybkiej migracji w kierunku anody, migrują one jednak w wraz z próbką ponieważ ich szybsza migracja spowodowała by powstanie luki jonowej i przerwanie pola elektrycznego. Po drugiej stronie układu z kolei obojętna ładunkowo glicyna nie powinna migrować, jednak w wyniku tego samego zjawiska zamyka strefę rozdziału podążając za próbką.
Z kolei wewnątrz pasa próbek dochodzi do uszeregowania cząsteczek względem ich ruchliwości. Odwołując się do wcześniej przytoczonego wzoru na prędkość migracji. Skoro prędkość jest taka sama, ruchliwość cząsteczek to stała dana (zadana dla cząsteczki i warunków), różna musi być także siła pola elektrycznego. Tworzy się jego gradient, powodujący uszeregowanie cząsteczek wg ich mobilności. Małe, silnie naładowane cząsteczki będą migrować z przodu (podobne do Cl) a duże o niższym ładunku będą z tyłu (Fig4). Przeprowadzając taki rozdział doprowadzamy do wyostrzenia stref i koncentracji cząsteczek. Izotachiforeza przeciwdziała dyfuzji, przez co szeroko stosuje się ją podczas elektroforezy nieciągłej/ elektroforezy DISC.
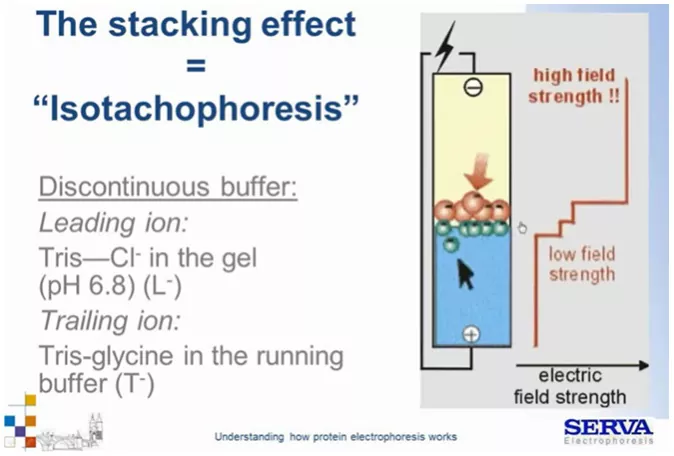
Fig 4.
Schemat ukazujący przebieg izotachioforezy. [źródło: Serva] [źródło: Serva]
Czemu podczas elektroforezy białek lepiej stosować żele poliakrylamidowe?
Dawniej stosowano żele agarozowe do rozdziału białek. Ponieważ jednak okazało się że poliakrylamidowe są lepsze, prawie całkowicie obojętne chemicznie i elektrochemicznie, wyparły one tańszy polimer.
Jak się okazuje żele agarozowe zawierają dużą ilość resztek agaropaktyny naturalnie występującej w agar agar. Związek ten nie sposób całkowicie usunąć i dochodzi do zjawiska silnej elektroendoosmozy. Gdy stosuje się mniejsze stężenie agarozy, w przypadku rozdziału większych cząsteczek jak kwasy nukleinowe, zjawisko to jest mniej problematyczne. Dlatego agar jest w tej aplikacji częściej używany niż podczas elektroforezy białek.
Żele poliakrylamidowe ponad to mają także inne zalety jak w tym kontekście. Zapewniają lepszą przezroczystość i elastyczność, mniejsza kruchość co ułatwia dalsze manipulacje i przeprowadzanie dalszych procedur.
Ponad to agaroza tworzy dość duże pory przez, które migrują białka, może zatem sprawdzać się podczas rozdziału większych cząsteczek ale nie można jej użyć do precyzyjniejszych rozdziałów.
Poliakrylamid z kolei umożliwia łatwe manipulowanie wielkością porów i dobieranie odpowiednich warunków stanowi dodatkowy, cenny walor separacyjny. Poprzez dobór odpowiednich proporcji akrylamidu i czynnika sieciującego można osiągnąć odpowiednią separację.
Przykładowo przy T 5% i C 3% (Total acrylamide/ Crosslinking) osiągniemy dość duże średnice porów około 5 nm.
Żel 1% agarozowy ma znacznie większą średnicę porów – 150 nm. (Fig5)
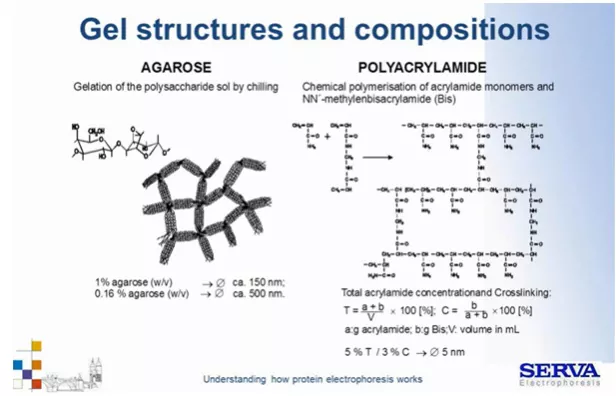
Fig 5.
Struktura porów żelu z agarozy i poliakrylamidu. [źródło: Serva]

Zobacz katalog produktów do SDS-PAGE
Czym jest elektroforeza DISC / nieciągła?
Technikę tę wprowadzili w 1964 roku Ornstein i Davis, którzy połączyli elektroforezę strefową oraz izotachioforezę. Konieczność jej opracowania była wymuszona stosowaniem w tamtych czasach nadal żelów agarozowych i skrobiowych o dużych porach, aby przeciwdziałać m.in. dysocjacji. Aktualnie wylewa się żele akrylamidowe jak wspomniana powyżej, jednak układ DISC nadal jest stosowany. Jaki problem rozwiązuje ta technika? Gdy klasycznie dla elektroforezy strefowej, umieścimy próbkę w dołkach i przyłożymy pole elektryczne cząsteczki zaczną szybko migrować w kierunku anody jeszcze w kieszonce. Chwilę później napotkają mechaniczną przeszkodę w postaci wąskich porów struktur żelu. Jeśli będzie on bardzo zatężony dojdzie do spowolnienia ich ruchu, koncentracji a w konsekwencji agregacji. Nie będą one prawidłowo wnikać w żel co będzie skutkować niewłaściwym rozdziałem i da się zaobserwować jako rozmyte prążki tzw. „smeary”.
Elektroforeza DISC zakłada takie wylanie żelu, aby w pierwszej fazie próbki łagodnie, efektywnie weszły w strukturę żelu. Możliwe jest to gdy podczas migracji najpierw będziemy prowadzili rozdział izotachioferetyczny, a dopiero później strefowy. Można tego dokonać odpowiednio wylewając żel – kolejnością przygotowania na spodzie żel rozdzielający (resolving gel) a powyżej żel tzw. zagęszczający (stucking gel), w którym przygotowujemy też dołki dla próbek. Oba żele różnią się zatężeniem akrylamidu i panującym pH. Żel zagęszczający na niższe pH (6,8) pozwalający glicynie na osiągnięcie obojętności chemicznej – to warunek konieczny, aby w układzie buforowym Tris-Glicyna doszło do izotachioforezy. W jej wyniku próbki wejdą w żel z tą samą prędkością flankowane przez glicynę, nie dopuszczając do agregacji białek. Kiedy cząsteczki podczas migracji napotkają bardziej zatężony żel rozdzielający zaczną zwalniać. Żel ten ma jednak wyższe pH (8,8-9,0) w wyniku czego glicyna zyskuje ładunek, a jako mała cząsteczka zaczyna migrować znacznie szybciej niż białka próbki. Pole elektryczne ze zmiennego przechodzi w jednorodne. Rozpoczyna się rozdział z różną prędkością zależnie od rozmiaru i ładunku cząsteczek (Fig6).
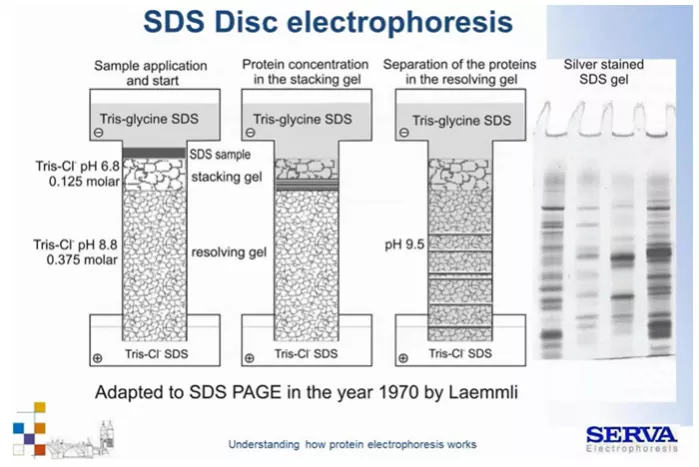
Fig 6.
Schemat elektroforezy nieciągłej / DISC. Szczegóły w tekście. [źródło: Serva]
Zobacz katalog produktów do SDS-PAGE
Czym jest elektroforeza SDS-PAGE?
Rozdziałowi SDS-PAGE, towarzyszy zaprowadzenie szczególnych warunków rozdziału. Stosuje się w niej detergent anionowy SDS czyli siarczan dodecylusodu- stąd nazwa.
Związek pokrywa cząsteczki białka nadając im ładunek ujemny i niwelując naładowanie grup aninowych obecnych w aminokwasach. W ten sposób eliminuje się rozdział białek ze względu na wypadkowy ładunek (Fig7). Na 1 g białka przypada ok 1,4 g SDS.
Ponad to stosując redukcję w połączeniu z denaturacją, sprawiamy, że cząsteczka staje się liniowa. Otrzymujemy zatem warunki, w których rozdział najbardziej liniowo zależy od parametru wielkości cząsteczek, czyli ich możliwości przeciskania się przez pory żelu. (Fig8)
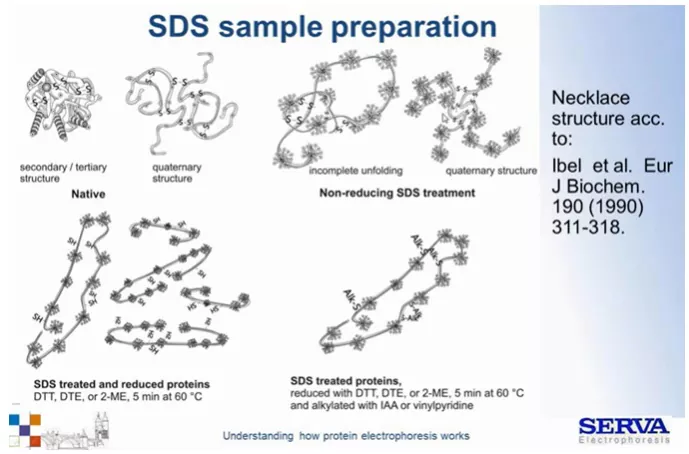
Fig 7.
Schematyczny obraz wpływu siarczanu dodecylu (SDS) na strukturę cząsteczki białka w warunkach redukujących i natywnych. W warunkach natywnych, cząsteczka jest pokryta ładunkiem ujemnym dzięki detergentowi, jej struktura jednak nie jest liniowa ze względu na obecność mostków disiarczkowych. Denaturacja/ gotowanie próbki w roztworze zawierającym silne reduktory – DTT (ditiotreitol) lub beta – merkaptoetanol. [źródło: Serva]
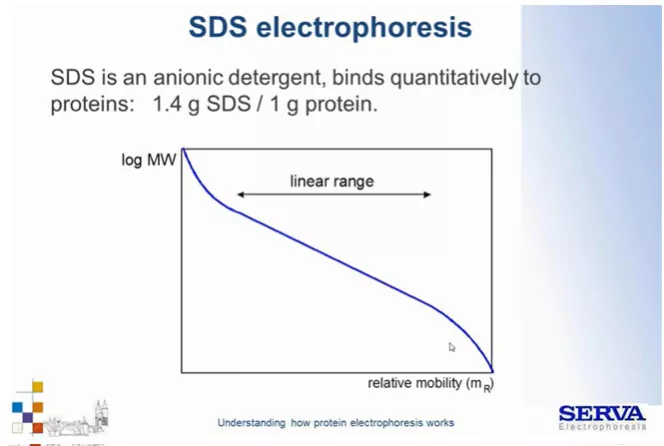
Fig 8.
Zależność migracji białek zależnie od ich wielkości w technice SDS-PAGE. Możemy zaobserwować, że im mniejsza cząsteczka tym szybciej migruje w polu elektrycznym. W technice SDS-PAGE zależność ta jest najbardziej liniowa, ze względu na ujednolicenie ładunku na całej długości rozdzielanego białka. [źródło: Serva]
Tracimy strukturę wyższego rzędu cząsteczki białka, ale zyskujemy możliwość lepszego rozpoznania i rozdzielenia cząsteczek. Denaturacji dokonuje się poprzez gotowanie próbki a redukcji poprzez dodanie czynników silnie redukujących takich jak popularny beta-merkaptoetanol lub DTT (ditiotreitol). Ponieważ związki te mają silne właściwości oksydoredukcyjne warto zadbać, aby były świeże i dodawane do buforu do próbek tuż przed tzw „gotowaniem” na termobloku. Aby zabezpieczyć uliniowienie cząsteczek przed odtworzeniem struktury wyższego rzędu (np. wiązań disiarczkowych) należy zalkalizować próbkę za pomocą jodoacetamidu lub winylopirydyny. Unikniemy powstawania rozmycia prążków na żelu.
Osobą, która połączyła elektroforezę DISC z SDS-PAGE był prof. Ulrich Laemmli, od którego nazwiska wziął nazwę bufor do próbek.
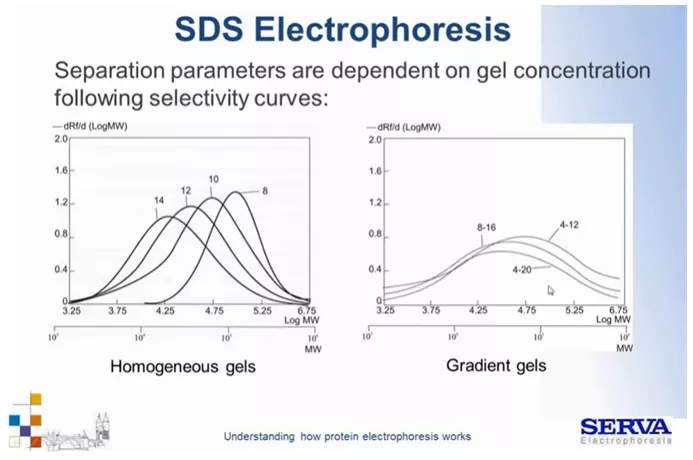
Fig 9.
Krzywe separacji cząsteczek zależnie od zatężenia żelu. [źródło: Serva]

Zobacz katalog produktów do SDS-PAGE
Odpowiednio manipulując zatężeniem, żelu możemy rozdzielać białka różnej wielkości (Fig9). Np. białka z zakresu wielkości 10^4 można rozdzielać na żelu 12-14% dla większych (10^5) lepiej stosować zatężenie 10-8%.
Możemy stosować żele o stałej procentowości akrylamidu lub gradientowe jeśli chcemy oznaczać białka z różnego zakresu wielkości. Analizę migracji ułatwiają tzw. drabinki białkowe / markery białkowe. Są to mieszaniny białek o znanej wielkości pozwalające na śledzenie postępu rozdziału. Warto pamiętać, że ze względu na dołączoną barwną grupę migrują one nieco wolniej niż swoje niewybarwione odpowiedniki. Na ich podstawie można szacować wielkość rozdzielanych białek (Fig10).
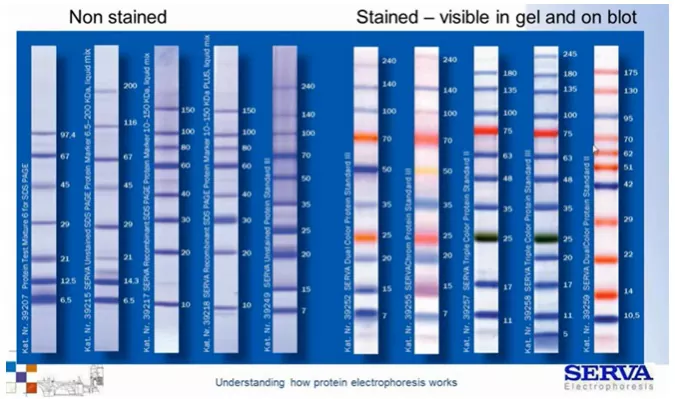
Fig 10.
Markery białkowe (drabinki białkowe) dostępne w naszej ofercie. [źródło: Serva]
Zobacz katalog produktów do SDS-PAGE
Czym jest elektroforeza natywna?
Elektroforeza pozwalająca na rozdział białek np. kompleksów bez niszczenia ich struktury 4-to i 3-cio rzędowej. Oznacza to, że nie denaturujemy ani nie redukujemy próbek a także nie stosujemy anionowych detergentów. Jeśli stosujemy rozdział nie ciągły, należy dokładnie wyznaczyć punkt izoelektryczny białka. Może się okazać, że znajduje się w okolicach 6,8 co oznacza brak możliwości rozdziału. Należy wtedy zastosować inny układ buforowy, wykorzystujący np. bufory kwaśne zawierające w swoim składzie CTAB i/lub 16-BAC. Powodujących z kolei uprotonowanie cząsteczek i uzyskanie wypadkowego dodatniego ładunku – elektroforezę przeprowadza się wtedy w kierunku anody. Taki rozdział jest szczególnie przydatny podczas elektroforezy dużych białek, silnie hydrofobowych np. glikoprotein błonowych.
Czym jest elektroforeza BN-PAGE (Blue Native-PAGE)?
Blue Native PAGE jest rodzajem elektroforezy natywnej, zmodyfikowanej w taki sposób, aby zapewnić cząsteczkom lepszą migrację w polu elektrycznym. Na klasyczny rozdział natywny wpływa zarówno ładunek wypadkowy cząsteczki jak oraz jej wielkość/nieregularność struktury. BN-PAGE wykorzystuje niebieski barwnik Coomassie Briliant Blue, który przykleja się do powierzchni hydrofobowych białek nadając im ujemny ładunek. W ten sposób można rozdzielać nawet bardzo duże białka. Barwnik przywierając do białek również je wybarwia umożliwiając śledzenie migracji w żelu (Fig11). Rozdział prowadzi się w neutralnym układzie buforowym, dzięki czemu składniki kompleksów białkowych można wyizolować i poddawać dalszej analizie.

Fig 11.
Zdjęcia etapów elektroforezy BN-PAGE oraz wynik takiego rozdziału z zabarwieniem pozostałym po barwniku Coomassie BB.
Zobacz katalog produktów do SDS-PAGE
Materiały do pobrania:





